
机器之心发布
机器之心编辑部
2024年诺贝尔化学奖将颁发给在结构生物学领域取得重大成果的David Baker团队和AlphaFold团队,刺激人工智能科学领域新的研究热潮。
近两年科学界颇具争议的一个命题是:“AlphaFold是否终结了结构生物学?”首先,AlphaFold等结构预测模型的训练数据来自X射线和冷冻电子显微镜(cryo-EM)。以etc为代表的传统结构分析方法。 其次,cryo-EM技术非常擅长分析蛋白质动力学,这是AlphaFold无法做到的。无独有偶,冷冻电子显微镜也获得了2017年诺贝尔化学奖。
那么,以AlphaFold为代表的人工智能技术能否辅助以冷冻电镜为代表的传统方法呢?两项诺奖级技术碰撞,能擦出怎样的火花?
字节跳动研究院提出了 AI for Science 模型,并被 Nature 子期刊收录
近日,字节跳动研究团队的研究人员提出了CryoSTAR方法,成功地将来自原子模型的结构应用于冷冻电镜实验数据的动态分析,为解决这一问题带来了新的思路和方法。 。与该成果相关的论文《CryoSTAR: Leveraging Structural Prior and Constraints for Cryo-EM Heterogeneous Reconstruction》已发表在《自然》杂志《NatureMethods》上。李忆来、周毅、袁静为共同作者,顾泉泉教授为通讯作者。

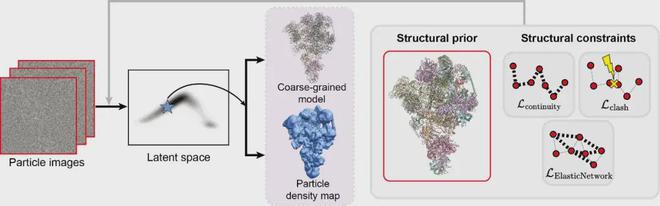
基于结构先验和约束的动态分析
CryoSTAR 是第一个成功地将蛋白质原子结构模式应用于冷冻电子显微镜实验数据的方法。该方法的主要特点是:
结合蛋白质结构的先验知识:CryoSTAR 方法创新性地使用原子模型信息作为结构正则化来解决冷冻电镜数据中的构象动力学问题,这与大多数以前仅从计算机视觉角度解决问题的方法不同。它需要一个初始原子模型作为参考,利用其结构信息来适当约束模型解析的构象动力学,从而缩小搜索空间,避免错误的分析结果,实现更好更快的收敛。输出两种模式的分析结果:该方法的独特之处在于可以同时输出粗粒度原子模型和密度图分析结果。这种多维度的结果输出有助于研究人员更全面地了解生物分子的结构。和动态过程。同时,密度图的输出结果还可以验证原子模型结果的置信度,检查粗粒度原子模型的动力学是否能够得到数据的充分支持。
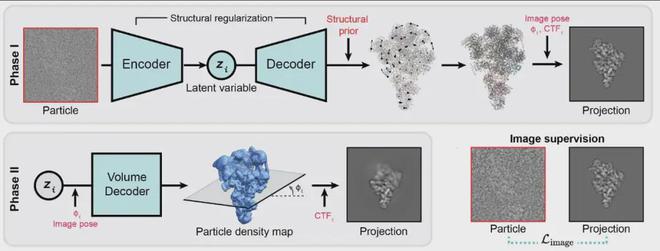
该方法分为两个阶段:
第一阶段通过受结构正则化约束的变分自动编码器(VAE)解析数据中的动态构象。 CryoSTAR 通过 Encoder 将构象动力学压缩为潜在变量;解码器将潜在变量解析为先前粗粒度原子模型的相应变形以拟合数据。它基于三个基本假设下的结构正则化:首先,同一条链上的两个相邻残基应始终保持连接,并受到连续损失函数的约束;其次,变形后预测的残差不能太接近,通过冲突损失函数来惩罚碰撞;第三,局部结构应尽可能刚性。通过从先验原子结构模型构建弹性网络并使用弹性网络损失函数,同时还使用自适应松弛方法来选择弹性网络,可以鼓励这种刚性。边缘被正则化以避免参考原子模型引起的过度偏差。第二阶段,训练密度图解码器以获得密度图模式的动态构象分析结果。 VAE训练完成后,提取所有图像对应的潜在变量并用于训练密度图解码器。密度图的细节优化受输入图像的约束,不受结构先验和正则化的直接影响,从而最小化结构先验对输出密度图的偏差,使得生成的密度图可以用于评估并验证粗粒度原子结构模型。 。
各种典型蛋白质数据的实验验证
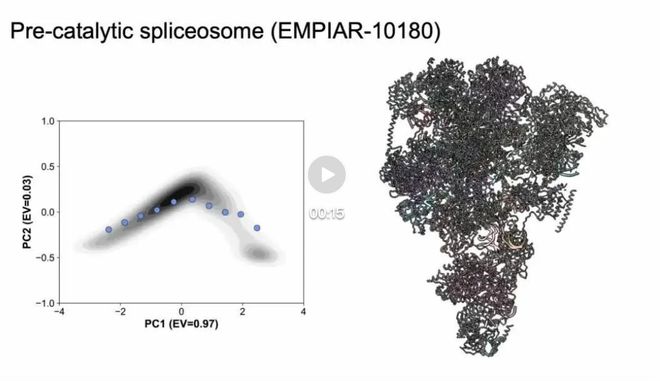
图3 EMPIAR-10180结果
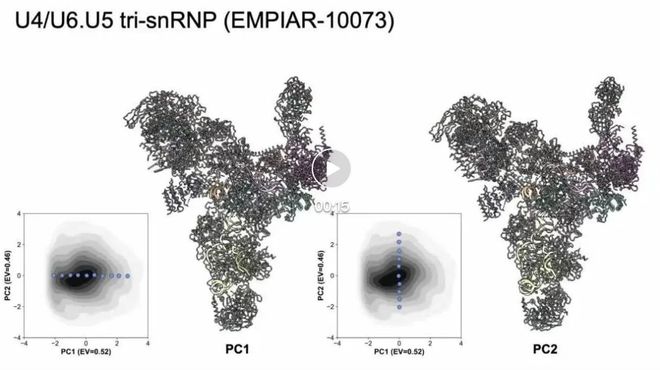
图 4 EMPIAR-10073 结果
大型复合物(残基数~10,000)
CryoSTAR 已在来自多个公开数据集 (EMPIAR) 的冷冻电镜实验数据上得到验证。对于酵母预催化B复合物剪接体(EMPIAR-10180),使用PDB:5NRL作为先验结构,cryoSTAR成功揭示了SF3b和解旋酶区域的构象变化。结果与其他方法一致,生成的粗粒度模型在电子密度图中得到了充分验证,显示出合理的运动模式。对于U4/U6.U5三核糖核蛋白(EMPIAR-10073),以PDB:5GAN为参考结构,cryoSTAR也分析了头部结构域的动态变化,结果与现有方法的发现一致。
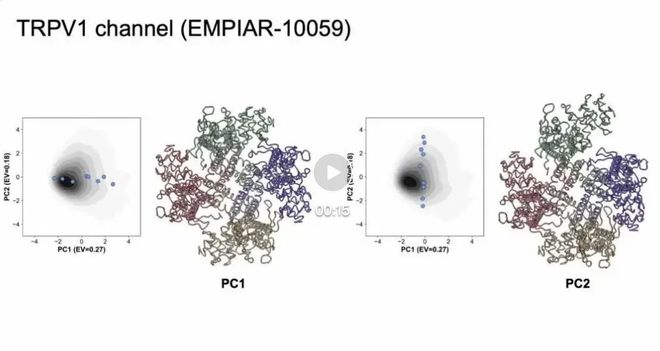
图 5 EMPIAR-10059 结果
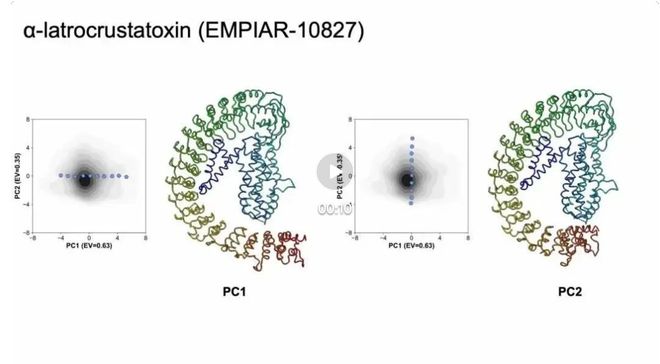
图 6 EMPIAR-10827 结果
膜蛋白(氨基酸数约2,000)
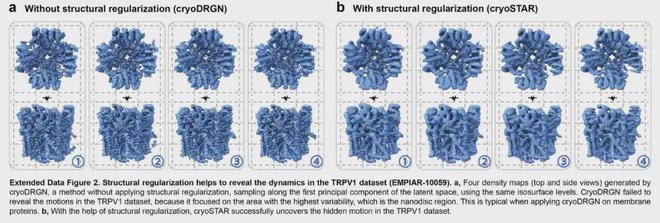
图7 CryoSTAR和CryoDRGN在EMPIAR-10059上的结果比较
对于TRPV1通道蛋白(EMPIAR-10059),使用PDB:7RQW作为参考先验原子模型,cryoSTAR发现该蛋白的外围可溶结构域存在微妙且平滑的运动,并且可以通过不同的主成分可视化每个亚基的运动。研究还表明,结构正则化在揭示膜蛋白动力学方面发挥着关键作用。相比之下,没有结构先验的方法(例如cryoDRGN)在膜蛋白的动态分析方面存在缺陷。
单链蛋白质(约 1,000 个氨基酸)
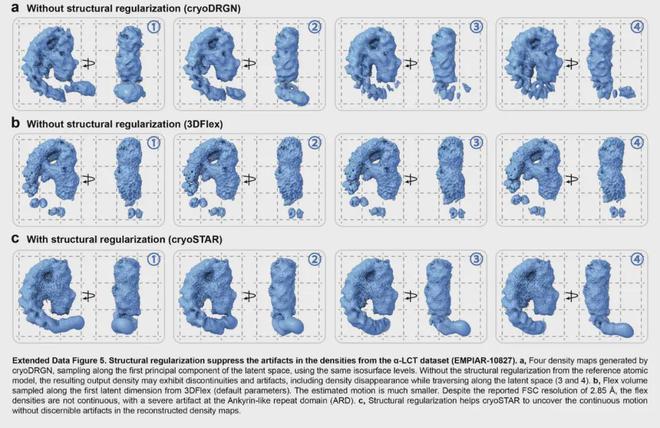
图8 CryoSTAR、CryoDRGN 和 3DFlex 在 EMPIAR-10059 上的结果比较
对于毒素蛋白α-LCT(EMPIAR-10827),cryoSTAR揭示了两种类型的运动,与离散3D分类发现的两种构象一致,生成的粗粒度原子模型合理,与密度图吻合良好。结构正则化对这种具有连续异质性的小蛋白质具有显着影响。与cryoDRGN、3DFlex等其他方法相比,cryoSTAR生成的密度图更加连续,可以有效避免密度图中的伪影。
摘要:使用跨模态数据解释自然现象
字节跳动研究院提出的结合结构先验的新方法(CryoSTAR)显着提升了冷冻电镜(Cryo-EM)在动态构象分析中的应用潜力。该创新方法以原子结构模式先验为约束,同时输出两种模式的解析结果,展现了其独特的技术优势,为科学研究打开了一扇新的大门。
这项研究在冷冻电镜动态分析领域的重要性不容忽视。传统的冷冻电镜方法由于分辨率和分析能力的限制,往往难以捕捉生物大分子复杂的动态变化。此次结构先验策略的引入,大大增强了动态构象分析的准确性和可靠性,使我们能够更深入地了解生物大分子的动态行为和机制。
这种方法的潜在价值在生命科学和制药领域尤其明显。生物大分子的动态分析是理解其功能机制的关键,例如蛋白质折叠、酶活性位点的变化、受体-配体结合模式等。这些动态过程与许多生理和病理现象密切相关,例如细胞信号传导、代谢调节和疾病机制。因此,准确分析这些动态过程不仅有助于揭示基本的生物学问题,而且为发现新的药物靶点和优化药物设计提供重要依据。在医药领域,该方法也具有广阔的应用前景。通过更高精度地捕捉蛋白质等生物大分子的动态变化,研究人员可以更有效地筛选潜在药物并设计更有针对性的药物分子,从而提高药物研发的效率和成功率。例如,该方法可以帮助分析与癌症、神经退行性疾病等重大疾病相关的蛋白质的动态构象变化,为新药的研发提供详细的分子水平信息。
总而言之,字节跳动研究院的这项创新研究不仅代表了冷冻电镜动态分析技术的最新进展,也为生命科学和制药领域带来了巨大的潜在价值。期待该方法未来能够在更多的研究和实际应用中得到验证和推广,为生命科学研究和药物研发带来新的突破和希望。
字节跳动研究院AI制药团队继续向AI for Science方向努力
字节跳动研究院AI制药团队致力于将人工智能技术应用于科学研究和药物开发。该团队在生成蛋白质设计、蛋白质构象预测、冷冻电镜分析等领域取得了显着的成果。
团队研究成果发表在Nature子刊、ICML、NeurIPS、ICLR等顶级学术会议上,得到学术界和工业界的广泛认可。
参考
[1] 郑Z,邓Y,薛D,等。结构知情语言模型是蛋白质设计师[C]//机器学习国际会议。 PMLR,2023:42317-42338。
[2] 王晓,郑志,叶芳,等。扩散语言模型是多功能蛋白质学习器 [C]。国际机器学习会议,2024 年。
[3] 王霞,郑志,叶芳,等。 DPLM-2:多模态扩散蛋白语言模型[J]. arXiv 预印本 arXiv:2410.13782, 2024。
[4] 周晓,薛东,陈瑞,等。通过直接基于能量的偏好优化进行抗原特异性抗体设计[C]。 NeurIPS,2024 年。
[5] 王Y,王L,沉Y,等。通过力引导 SE 生成蛋白质构象 (3) 扩散模型 [C]。国际机器学习会议,2024 年。
[6] 李Y,周Y,袁J,等。 CryoSTAR:利用结构先验和约束进行冷冻电镜异质重建 [J]。自然方法,2024:1-9。
[7] 周Y,李Y,袁J,等。 CryoFM:基于流的冷冻电镜密度基础模型。 arXiv 预印本 arXiv:2410.08631, 2024。
文本视频链接:#rd
本文采摘于网络,不代表本站立场,转载联系作者并注明出处:http://mjgaz.cn/fenxiang/271406.html
